

Wady wrodzone i zespoły genetyczne stanowią jedno z największych wyzwań współczesnej pediatrii. Różnorodność objawów klinicznych, rzadkie występowanie i wciąż nie do końca poznane mechanizmy odpowiedzialne za powstawanie wad wrodzonych powodują, że ich diagnostyka jest procesem długotrwałym i wieloetapowym. Jednym z rzadkich zaburzeń o podłożu genetycznym, ujawniającym się już w okresie noworodkowym jest zespół Wolfa-Hirschhorna. Jak objawia się to schorzenie oraz na czym polega jego leczenie?
Co to jest zespół Wolfa-Hirschhorna?
Zespół Wolfa-Hirschhorna jest to ciężkie zaburzenie rozwojowe o podłożu genetycznym, charakteryzujące się obecnością wad twarzoczaszki, upośledzeniem wzrostu, opóźnieniem rozwoju psychoruchowego oraz częstym występowaniem drgawek. Częstość występowania zespołu Wolfa-Hirschhorna w populacji światowej szacowana jest na około 1:50 000 żywych urodzeń. Schorzenie to dwa razy częściej dotyka płci żeńskiej, a jego objawy zwykle występują już u noworodków.
Objawy zespołu Wolfa-Hirschhorna

Najbardziej typowy dla tej jednostki chorobowej jest specyficzny wygląd twarzy pacjentów, opisywany jako przypominający “hełm greckiego wojownika”. Na charakterystyczny fenotyp zespołu Wolfa-Hirschhorna składają się:
- małogłowie – wymiar obwodu głowy jest znacznie mniejszy od ustalonych dla wieku i płci wartości referencyjnych;
- szeroka nasada nosa, sięgająca aż do czoła;
- wysokie czoło;
- hiperteloryzm – zbyt szerokie rozstawienie oczodołów i gałek ocznych;
- wysokie łuki brwiowe;
- zmarszczki nakątne oczu – fałdy skórne ciągnące się od górnej do dolnej powieki. Pokrywają one przyśrodkowy kąt szpary powiekowej;
- krótka rynienka podnosowa – jest to fizjologiczne zagłębienie biegnące od przegrody nosa do brzegu wargi górnej;
- kąciki ust skierowane ku dołowi;
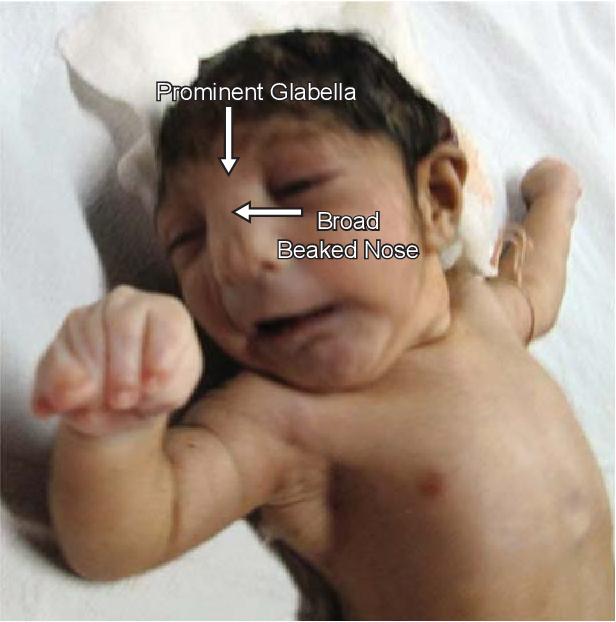
- niedorozwój żuchwy;
- wąskie, wysoko wysklepione podniebienie (określane jako podniebienie gotyckie);
- małe, nisko osadzone uszy;
- u części pacjentów występuje rozszczep wargi i/lub podniebienia.
Cechy te są bardzo są charakterystyczne i pozwalają na postawienie wstępnego rozpoznania choroby już w okresie noworodkowym. Należy jednak podkreślić, że objawy zespołu Wolfa-Hirschhorna mogą być bardzo różnorodne, a ich nasilenie jest zmienne wśród pacjentów. Różnorodność objawów zależy od ilości utraconego w wyniku delecji materiału genetycznego u konkretnego pacjenta (podłoże genetyczne choroby zostało szeroko omówione w dalszej części artykułu).
Często już podczas ciąży, w badaniu ultrasonograficznym (USG) stwierdza się ograniczenie wzrastania wewnątrzmacicznego – płód jest zbyt mały w stosunku do wieku ciążowego. Noworodki cechuje niska masa urodzeniowa, mają one także problemy z przybieraniem na wadze. Trudności w karmieniu i brak przyrostu masy ciała częściowo spowodowane są niedorozwojem mięśni oraz obniżonym napięciem mięśniowym (hipotonią), przez co noworodki i niemowlęta nie wykształcają prawidłowego odruchu ssania pokarmu.
Pacjenci z zespołem Wolfa-Hirschhorna prezentują znaczne opóźnienie rozwoju psychoruchowego – większość chorych nie uzyskuje kontroli nad zwieraczami (oddają bezwiednie kał i mocz), nie rozwijają oni także umiejętności samodzielnego jedzenia ani ubierania. Mniej niż połowa chorych wykształca umiejętność chodzenia. Pacjenci wykazują niepełnosprawność intelektualną w stopniu umiarkowanym do ciężkiego – w znacznej większość przypadków nie potrafią oni mówić, a komunikacja z otoczeniem ogranicza się do wydawania kilkusylabowych dźwięków.
Większość pacjentów cierpi na napady drgawek, których występowanie często jest prowokowane przez gorączkę. U około połowy chorych ataki padaczkowe ustępują samoistnie w dzieciństwie. Chorzy z zespołem Wolfa-Hirschhorna często mają strukturalne (anatomiczne) uszkodzenia ośrodkowego układu nerwowego, które mogą odpowiadać za pojawianie się napadów drgawek, a także powodować niepełnosprawność intelektualną.
Inne problemy zdrowotne, z którymi muszą zmagać się pacjenci z zespołem Wolfa-Hirschhorna, to:
- nieprawidłowości w budowie układu kostno-szkieletowego – skolioza, patologiczna kifoza, dodatkowe lub zrośnięte żebra, stopa końsko-szpotawa;
- wrodzone wady serca oraz oczu (zez);
- ubytek słuchu;
- braki w uzębieniu;
- nawracające zapalenia płuc, ucha środkowego i układu moczowego, wynikające z niedoboru przeciwciał.
Zaburzenia te dotyczą jedynie części chorych, a ich pojawienie się jest uzależnione od tego, jak duży ubytek materiału genetycznego (wynikający z delecji) występuje u danego pacjenta.
*fenotyp – zespół cech morfologicznych i fizjologicznych organizmu, które są możliwe do zaobserwowania;
*skolioza – skrzywienie boczne kręgosłupa;
*patologiczna kifoza – nadmierne wygięcie kręgosłupa do tyłu w odcinku szyjnym, piersiowym lub lędźwiowym;
*stopa końsko-szpotawa – wrodzona wada stóp polegająca na przywiedzeniu przodostopia i utrwalonym zgięciu podeszwowym stopy.
Przyczyny zespołu Wolfa-Hirschhorna
Jak już wspomniano, zespół Wolfa-Hirschhorna jest schorzeniem o podłożu genetycznym – jego przyczyną jest częściowa delecja w obrębie krótkiego ramienia 4 chromosomu. Delecja jest to rodzaj mutacji genetycznej, polegający na utracie fragmentu chromosomu, co wiąże się z nieodwracalną utratą genów znajdujących się w tym obszarze. U pacjentów z zespołem Wolfa-Hirschhorna dochodzi do utraty takich genów, jak NSD2, LETM1, WHSC1, MSX1. Odgrywają one ważną rolę we wczesnym rozwoju płodowym, ale określenie dokładnej funkcji każdego z genów wymaga przeprowadzenia dalszych badań. Postuluje się, że:
- gen NSD2 prawdopodobnie odpowiada za prawidłowy rozwój twarzy, a jego utrata powoduje charakterystyczny wygląd twarzy pacjentów z zespołem Wolfa-Hirschhorna;
- utrata genu LETM1 może wiązać się z występowaniem napadów padaczkowych;
- utrata genu MSX1 prawdopodobnie odpowiada za zwiększone ryzyko występowania rozszczepu wargi i podniebienia.
Większe delecje (obejmujące utratę dużej liczby genów), związane są z wyższym ryzykiem występowania wad serca i rozszczepu podniebienia.
Czy wiesz że:
mediana przeżycia pacjentów, u których zespół Wolfa-Hirschhorna wystąpił na skutek mutacji de novo wynosi powyżej 34 lat, a u chorych z translokacją niezrównoważoną - powyżej 18 lat?
W około 90% przypadków zespół Wolfa-Hirschhorna nie jest dziedziczny (nie występuje w rodzinie w poprzednich pokoleniach) i powstaje na skutek mutacji de novo – do delecji dochodzi dopiero podczas zapłodnienia, czyli połączenia plemnika i komórki jajowej.
Znacznie rzadziej, schorzenie to może zostać odziedziczone od rodziców, w przypadku gdy przynajmniej jedno z nich jest nosicielem tzw. translokacji zrównoważonej. Jest to mutacja polegająca na “oderwaniu się” i zamianie miejscami fragmentów dwóch różnych chromosomów. W tej sytuacji całkowita ilość materiału genetycznego nie zmienia się, lecz jest on rozmieszczony nieprawidłowo. Stan ten zwykle nie ma żadnych następstw klinicznych i nosiciel tego rodzaju mutacji nie jest jej świadomy, może ona jednak spowodować, że w kolejnym pokoleniu dojdzie do translokacji niezrównoważonej. Jest to sytuacja, w której występuje dodatkowy fragment jednego chromosomu i/lub brakuje fragmentu innego chromosomu. Wystąpienie translokacji niezrównoważonej u dziecka prowadzi do wystąpienia różnego rodzaju zaburzeń i objawów klinicznych, których rodzaj i nasilenie uzależniony jest od tego, jak dużo materiału genetycznego ubyło lub zostało dodane oraz jakich chromosomów zmiany te dotyczą.
Diagnostyka zespołu Wolfa-Hirschhorna
Ze względu na charakterystyczny wygląd twarzy pacjentów, wstępne rozpoznanie choroby zwykle stawiane jest na podstawie obrazu klinicznego krótko po narodzinach dziecka. O ostatecznym ustaleniu rozpoznania zespołu Wolfa-Hirschhorna rozstrzyga badanie cytogenetyczne, polegające na analizie liczbowej i strukturalnej chromosomów. Pozwala ono na wykrycie utraty fragmentu ramienia krótkiego chromosomu 4, co jest zmianą typową dla tego schorzenia. Modyfikacją klasycznego badania cytogenetycznego jest badanie metodą FISH (fluorescencyjna hybrydyzacja in situ), które jest najdokładniejszym sposobem potwierdzenia zespołu Wolfa-Hirschhorna. Badanie to ma na celu wykrycie w materiale genetycznym określonej sekwencji DNA przy pomocy specjalnych fluorescencyjnych sond DNA, które w przypadku zidentyfikowania poszukiwanego fragmentu zaczynają świecić. Badanie metodą FISH wykonuje się, gdy za pomocą klasycznego badania cytogenetycznego nie udaje się ustalić dokładnego rodzaju lub miejsca mutacji. Technika FISH umożliwia wykrycie u pacjenta zarówno delecji, jak i translokacji niezrównoważonej.
Jak leczyć zespół Wolfa-Hirschhorna?
Jak dotąd nie opracowano skutecznego leczenia przyczynowego zespołu Wolfa-Hirschhorna, a podstawą terapii pozostaje niwelowanie dokuczliwych objawów towarzyszących chorobie. Leczenie objawowe polega na:
- zapobieganiu napadom drgawek (stosowanie leków przeciwpadaczkowych np. kwasu walproinowego, etosuksymidu);
- intensywnej rehabilitacji;
- opiece psychologa i neurologopedy.
Wyżej wymienione sposoby terapii stosowane są w celu poprawy komfortu życia pacjentów z zespołem Wolfa-Hirschhorna, ale nie przedłużają go.
Zapamiętaj:
Zespół Wolfa-Hirschhorna jest to ciężkie zaburzenie rozwojowe o podłożu genetycznym, charakteryzujące się obecnością wad twarzoczaszki, upośledzeniem wzrostu, opóźnieniem rozwoju psychoruchowego oraz częstym występowaniem drgawek.
Współwystępowanie wad wrodzonych narządów, takich jak np. wady serca, oczu czy też nieprawidłowości układu kostno-stawowego, wymaga leczenia przyczynowego – operacji kardiochirurgicznych, zabiegów okulistycznych oraz ortopedycznych. Najbliższa rodzina pacjenta powinna zostać objęta opieką poradni genetycznej. Pacjenci z zespołem Wolfa-Hirschhorna przez całe życie muszą pozostawać pod kontrolą wielospecjalistycznego zespołu, w skład którego wchodzą pediatrzy, genetycy kliniczni, neurolodzy, okuliści, laryngolodzy, kardiolodzy, ortopedzi, psychologowie i fizjoterapeuci.
Zespół Wolfa-Hirschhorna jest rzadkim schorzeniem o podłożu genetycznym, występującym częściej u płci żeńskiej. Pacjentów z tym rozpoznaniem cechuje specyficzny wygląd twarzy (przypominający hełm greckiego wojownika), obniżenie napięcia mięśniowego, znaczne opóźnienie rozwoju psychoruchowego oraz występowanie napadów drgawek. Przyczyną schorzenia jest delecja w obrębie ramienia krótkiego chromosomu 4, a diagnoza stawiana jest na podstawie obrazu klinicznego oraz wyników badania cytogenetycznego. Na dzień dzisiejszy nie ma możliwości leczenia przyczynowego tego schorzenia, a komfort życia pacjentów jest poprawiany dzięki terapii objawowej i intensywnej rehabilitacji.