

Mitochondria są to organella komórkowe (otoczone błoną, wyspecjalizowane struktury) potocznie określane mianem “wewnętrznych elektrowni”- odpowiadają one za zaopatrywanie komórek naszego ciała w energię, która powstaje w procesie oddychania komórkowego, zachodzącego właśnie w mitochondriach. Proces ten jest wieloetapowy i polega na utlenianiu glukozy do dwutlenku węgla, w wyniku czego uwalniana jest energia, która następnie jest wykorzystywana do procesów życiowych komórki. Im większe jest zapotrzebowanie energetyczne komórki, tym bogatsza jest ona w mitochondria. Najwięcej mitochondriów zawierają komórki nerwowe oraz mięśniowe. Choroby mitochondrialne to uwarunkowana genetycznie grupa schorzeń, które prowadzą do zaburzeń funkcji mitochondriów, skutkujących niedostatecznym zaopatrzeniem komórek w energię. Jednym ze schorzeń związanych nieprawidłowym funkcjonowaniem mitochondriów jest zespół Leigha – jak się on objawia oraz jakie jest rokowanie u pacjentów z tym rozpoznaniem?
Co to jest zespół Leigha?
Zespół Leigha jest to rzadka choroba neurodegeneracyjna o podłożu genetycznym, zaliczana do grupy chorób mitochondrialnych. Powstaje ona na skutek mutacji wielu różnych genów kodujących enzymy mitochondrialne, które odpowiadają za prawidłowy przebieg procesu oddychania komórkowego. W przebiegu schorzenia dochodzi do zaburzeń oddychania komórkowego, co prowadzi do niedostatecznego zaopatrzenia tkanek w energię. Najbardziej podatny na nieprawidłową czynność mitochondriów jest ośrodkowy układ nerwowy, serce oraz mięśnie szkieletowe i to w tych narządach występują pierwsze objawy schorzeń mitochondrialnych. Częstość występowania zespołu Leigha szacuje się na 1: 36 000 żywo urodzonych dzieci.
Zespół Leigha – objawy

Zespół Leigha jest schorzeniem wrodzonym, ale objawy kliniczne nie są widoczne tuż po urodzeniu. Pierwsze symptomy z reguły pojawiają się między 3 miesiącem a 2 rokiem życia dziecka. W bardzo rzadkich przypadkach pierwsze dolegliwości mogą pojawić się u pacjenta w wieku nastoletnim, jednak opisano zaledwie kilkoro takich pacjentów.
Do najwcześniejszych objawów chorobowych obserwowanych u pacjentów z zespołem Leigha zaliczamy:
- zbyt słaby odruch ssania;
- problemy z trzymaniem główki;
- utratę apetytu;
- wymioty;
- drażliwość, pobudzenie, płaczliwość.
Symptomy te są bardzo niespecyficzne i mogą występować w przebiegu wielu różnych schorzeń, typowych dla populacji pediatrycznej. W następnych etapach choroby do tych objawów dołączają się zaburzenia neurologiczne oraz osłabienie mięśni, które klinicznie manifestują się:
- regresem rozwoju psychoruchowego;
- napadami drgawek;
- obniżonym napięciem mięśniowym (wiotkością mięśni);
- postępująca ataksją – zaburzeniem koordynacji ruchów, niezbornością ruchów;
- oczopląsem – mimowolnymi, niekontrolowanymi ruchami gałek ocznych;
- utratą słuchu;
- osłabieniem siły mięśniowej.
W wyniku dysfunkcji mitochondriów i niedostatecznego zaopatrzenia komórek nerwowych w energię dochodzi do powstawania uszkodzeń (ognisk martwicy) w obrębie ośrodkowego układu nerwowego, które manifestują się wyżej wymienionymi objawami klinicznymi. Można je uwidocznić w rezonansie magnetycznym (MR) głowy. Objawy kliniczne w dużej mierze uzależnione są od dokładnej lokalizacji zmian martwiczych w mózgu.
Zespół Leigha może również przebiegać z zaburzeniami ze strony innych narządów – głównie serca, wątroby i nerek. Tak liczne dysfunkcje w obrębie kluczowych dla życia narządów powodują, że nawet jeżeli we wczesnym dzieciństwie pacjenci z zespołem Leigha rozwijali się prawidłowo i np. nauczyli się chodzić, to w przebiegu choroby bezpowrotnie tracą te umiejętności i stają się całkowicie uzależnieni od pomocy drugiego człowieka. U chorych z zespołem Leigha obserwuje się tendencję do występowania naprzemiennych zaostrzeń (pogorszenia stanu zdrowia) i remisji. Po okresach względnej poprawy następuje nagła dekompensacja, po której chory już nie powraca do poprzedniego stanu klinicznego.
Czy wiesz że:
w Polsce działa stowarzyszenie “Mali Bohaterowie”, które zrzesza rodziny dzieci z zespołem Leigha?
Stan zdrowia pacjentów stopniowo pogarsza się- do wyżej wymienionych objawów dołączają się zaburzenia wynikające z uszkodzenia pnia mózgu, takie jak dysfagia (zaburzenia połykania) czy trudności z oddychaniem. Ze względu na to, że do rozwoju zespołu Leigha może prowadzić wiele różnych mutacji genetycznych, objawy prezentowane przez chorych oraz ich ogólny stan kliniczny mogą się bardzo różnić między pacjentami.
*pień mózgu – część ośrodkowego układu nerwowego, w skład której wchodzą śródmózgowie, most oraz rdzeń przedłużony. W pniu mózgu zlokalizowane są ośrodki odpowiedzialne za kontrolowanie podstawowych procesów życiowych, takich jak krążenie krwi, oddychanie czy połykanie. Uszkodzenie którejkolwiek ze struktur wchodzących w skład pnia mózgu wiąże się z bardzo dużym ryzykiem zgonu, głównie na skutek ustania procesów krążenia i oddychania.
Zespół Leigha – przyczyny
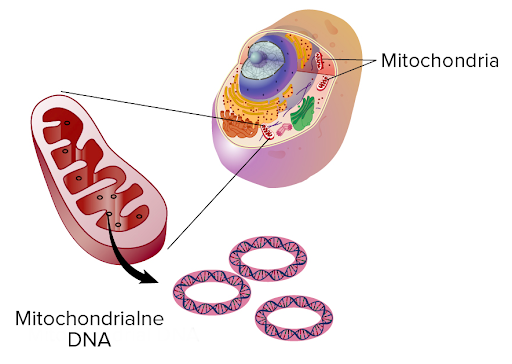
Jak już wspomniano, zespół Leigha jest schorzeniem dziedzicznym, spowodowanym mutacją genetyczną. Należy dodać, że zespół ten jest chorobą bardzo heterogenną genetycznie – oznacza to, że do jej rozwoju mogą prowadzić zarówno mutacje w jądrowym DNA, jak i w DNA znajdującym się w mitochondriach. Informacja genetyczna człowieka zawarta jest w dwóch typach DNA- jądrowym oraz mitochondrialnym. DNA jądrowe dziedziczone jest od obojga biologicznych rodziców, zaś DNA mitochondrialne- wyłącznie po matce dziecka. Przyczyna tego zjawiska została wyjaśniona w dalszej części artykułu.
Jak dotąd zidentyfikowano ponad 30 mutacji, które mogą prowadzić do rozwoju zespołu Leigha. Najogólniej mówiąc, zespół ten może być spowodowany zaburzeniem któregokolwiek z mitochondrialnych kompleksów łańcuchów oddechowych. Mitochondrialny łańcuch oddechowy jest to zespół enzymów (kompleksów białkowych oznaczonych cyframi I-IV), zlokalizowanych na wewnętrznej błonie mitochondrialnej i uczestniczących w skomplikowanych przemianach biochemicznych, których finalnym efektem jest wytworzenie energii niezbędnej do procesów życiowych komórki.
Do najczęstszych mutacji genetycznych stwierdzanych w zespole Leigha zaliczamy defekty następujących genów:
- SURF1;
- MTATP6;
- PDHA1.
W warunkach fizjologicznych białka będące produktami tych genów wchodzą w skład kompleksów (enzymów) łańcucha oddechowego. Mutacje enzymów mitochondrialnych prowadzą do zaburzeń procesów oddychania komórkowego, co skutkuje niedostatecznym zaopatrzeniem komórek w energię – najbardziej zauważalne jest to w komórkach o dużym zapotrzebowaniu energetycznym, takich jak komórki nerwowe i mięśniowe. Zejściowym efektem tych dysfunkcji jest stopniowe, nieodwracalne obumieranie komórek, co manifestuje się szeregiem objawów klinicznych, w tym głównie ze strony układu nerwowego i mięśni.
Źródła: http://biologiabeztajemnicsp15.blogspot.com/2018/09/komorka.html
https://pl.khanacademy.org/science/ap-biology/heredity/non-mendelian-genetics/a/mitochondrial-and-chloroplast-dna-inheritance
Różne są także sposoby dziedziczenia tego schorzenia – w większości przypadków zespół Leigha dziedziczony jest w sposób autosomalny (niezależny od płci) recesywny. Oznacza to, że do wystąpienia choroby u dziecka konieczne jest odziedziczenie dwóch zmutowanych kopii genu, od obojga z rodziców. W części przypadków jednak zespół Leigha może być dziedziczony w sposób:
- mitochondrialny (odmatczyny) – ten rodzaj dziedziczenia zespołu Leigha ma miejsce wówczas, gdy mutacji uległy geny wchodzące w skład DNA zawartego w mitochondriach (mtDNA), które jest przekazywane potomstwu wyłącznie przez matkę. Wynika to z faktu, że plemniki zawierają bardzo niewielką ilość mitochondrialnego DNA, które nie dostaje się do komórki jajowej podczas procesu zapłodnienia. Oznacza to, że choroby spowodowane mutacjami w DNA mitochondrialnym są przekazywane potomstwu wyłącznie przez kobiety;
- recesywny, sprzężony z chromosomem płciowym X – każdy człowiek posiada parę chromosomów płciowych. W przypadku kobiet są to dwa chromosomy X (jeden od matki, a drugi od ojca), mężczyźni zaś posiadają chromosom X od matki oraz chromosom Y ojca. Chromosom X jest znacznie większy od chromosomu Y i zawiera dużo więcej genów. Różnica ta powoduje, że jeśli kobiety odziedziczą jedną zmutowaną kopię genu związanego z chromosomem X, to nie dojdzie u nich do rozwoju choroby, ze względu na posiadanie drugiej, prawidłowej kopii tego samego genu na drugim chromosomie X, która niejako “zrównoważy” obecność zmienionej kopii. W tym przypadku kobieta jest nosicielką choroby- nie prezentuje objawów choroby, ale może przekazać zmutowaną kopię genu swoim dzieciom. U mężczyzn sytuacja przedstawia się inaczej- mają oni tylko jeden chromosom X, dlatego jeśli jeden z genów na chromosomie X zawiera mutację, to nie mają oni drugiej kopii tego samego genu, która mogłaby zrównoważyć wadliwą kopię. Oznacza to, że wystąpią u nich objawy choroby.
Genetyczna przyczyna wielu przypadków zespołu Leigha pozostaje wciąż nieznana. Rodziny, w których zdiagnozowano przypadek zespołu Leigha, powinny zostać objęte opieką poradni genetycznej.
Zespół Leigha – leczenie
Na chwilę obecną nie odkryto terapii, która umożliwiłaby trwałe wyleczenie zespołu Leigha. Ze względu na fakt, że w chorobach mitochondrialnych często dochodzi do nadprodukcji wolnych rodników, u chorych wspomagająco stosuje się witaminę B1- tiaminę, witaminę B2- ryboflawinę oraz koenzym Q10, które wykazują działanie przeciwutleniające- chronią komórki, w tym mitochondria, przed stresem oksydacyjnym (stanem zakłóconej równowagi między szkodliwymi wolnymi rodnikami a zdolnością ich neutralizacji przez organizm). Skuteczność wyżej wymienionych preparatów uzależniona jest od rodzaju defektu genetycznego występującego u konkretnego pacjenta. U pacjentów z zespołem Leigha, u których dodatkowo stwierdzono niedobór kompleksu enzymów dehydrogenazy pirogronianowej zaleca się stosowanie diety wysokotłuszczowej, ubogiej w węglowodany. Dieta ta wykazuje skuteczność u tej grupy chorych, ponieważ niedobór dehydrogenazy pirogronianowej wiąże się z zaburzeniami metabolizmu węglowodanów (cukrów). Ograniczenie spożycia cukrów oraz wprowadzenie do diety większej ilości tłuszczów, które organizm zaczyna wykorzystywać jako główne źródło energii, znacznie poprawia stan ogólny pacjentów i wpływa na zmniejszenie nasilenia dokuczliwych objawów klinicznych.
Zapamiętaj:
Pierwsze symptomy zespołu Leigha z reguły pojawiają się między 3 miesiącem a 2 rokiem życia dziecka.
Duże nadzieje na poprawę losu chorych i spowolnienie postępu choroby wiązane są obecnie z badaniami klinicznymi nad nowym lekiem zwanym EPI-743. Swym działaniem przypomina on koenzym Q10, który fizjologicznie występuje w mitochondriach. Wykazuje właściwości przeciwutleniające oraz zwiększa efektywność procesów oddychania komórkowego, przez co w mitochondriach produkowane są większe ilości energii. W przeprowadzonych dotychczas badaniach klinicznych nowy lek znacznie łagodził objawy choroby oraz poprawiał komfort życia chorych, jednak z uwagi na podłoże genetyczne zespołu Leigha nie umożliwia on trwałego wyleczenia.
Ze względu na mnogość problemów zdrowotnych towarzyszących zespołowi Leigha, opieka nad chorymi wymaga zaangażowania zespołu wielu specjalistów, w skład którego wchodzą pediatrzy, kardiolodzy, neurolodzy, otorynolaryngolodzy, okuliści, specjaliści medycyny paliatywnej, rehabilitanci oraz dietetycy. Pacjenci wymagają kompleksowego i systematycznego kontrolowania stanu zdrowia oraz w zależności od potrzeb, modyfikacji terapii.
*dehydrogenaza pirogronianowa – zespół trzech enzymów zaliczanych do kompleksu łańcucha oddechowego.
Ile żyją dzieci z zespołem Leigha?
Rokowanie w przypadku rozpoznania zespołu Leigha jest niepomyślne, a długość życia uzależniona jest od rodzaju defektu genetycznego leżącego u podłoża zespołu u konkretnego pacjenta. Pacjenci, u których stwierdzono brak mitochondrialnego kompleksu IV łańcucha oddechowego oraz osoby z niedoborem dehydrogenazy pirogronianowej mają zwykle najgorsze rokowanie i umierają w ciągu kilku pierwszych lat życia. Osoby z częściowymi niedoborami enzymów mitochondrialnych mają zwykle nieco lepsze rokowanie i dożywają do około 7 roku życia. Przyczyną zgonu jest najczęściej niewydolność oddechowa.
Choroby mitochondrialne, ze względu na swe rzadkie występowanie, różnorodne manifestacje kliniczne, złożone podłoże genetyczne oraz trudności diagnostyczne wciąż stanowią ogromne wyzwanie dla współczesnej medycyny. Rokowanie w chorobach mitochondrialnych, w tym zespole Leigha jest niekorzystne, a przebieg kliniczny tych schorzeń bywa bardzo nieprzewidywalny. W przypadku ich rozpoznania poradnictwem genetycznym oraz opieką specjalistyczną należy objąć całą najbliższą rodzinę pacjenta.