

Kształt i wymiary czaszki nie są jednakowe przez całe życie – jak każda kość w organizmie, kości czaszki powiększają się wraz ze wzrostem dziecka. Powiększanie się wymiarów czaszki w wieku dziecięcym jest możliwe dzięki obecności szwów czaszkowych-włóknistych przestrzeni pomiędzy kośćmi czaszki niemowlęcia. Funkcją szwów jest umożliwienie zwiększania się rozmiarów czaszki, proporcjonalnie do wzrostu rozwijającego się mózgu, którego objętość rośnie aż 3-krotnie w ciągu pierwszych lat życia dziecka. Przedwczesne zarośnięcie szwów czaszkowych stanowi podłoże jednostki chorobowej zwanej zespołem Crouzona. Jak objawia się zespół Crouzona oraz na czym polega jego leczenie?
Czym jest zespół Crouzona?
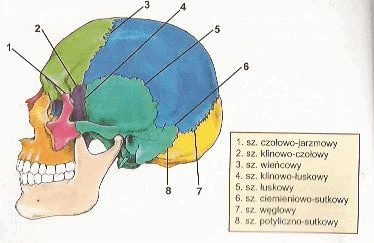
Zespół Crouzona jest to zaburzenie genetyczne objawiające się występowaniem szeregu wad twarzoczaszki. Podłożem występowania objawów chorobowych w zespole Crouzona jest przedwczesne zarastanie szwów czaszkowych- zjawisko to określane jest mianem kraniosynostozy. Do najważniejszych szwów czaszkowych należą:
- szew strzałkowy;
- szew wieńcowy;
- szew węgłowy;
- szew łuskowy.
W warunkach fizjologicznych zarastanie szwów ma ściśle określoną kolejność, a czas trwania tego procesu jest kwestią osobniczą, jednak szacuje się, że kończy się on około 5 roku życia. W przypadku zespołu Crouzona kraniosynostoza rozpoczyna się tuż po urodzeniu, a kończy około 2-3 roku życia. Zdarza się, że zarośnięcie szwów czaszkowych jest stwierdzane już podczas ciążowych badań ultrasonograficznych Przedwczesne zarośnięcie szwów czaszkowych zaburza naturalny wzrost i rozwój kości czaszki, powodując zniekształcenia głowy i twarzy, a przede wszystkim uniemożliwia proporcjonalne powiększanie objętości mózgu. Zespół Crouzona jest zaburzeniem rzadkim- jego częstość szacuje się na około 16 : 1 000 000 żywych urodzeń, jednakowo często dotyczy on płci żeńskiej i męskiej.
Zespół Crouzona należy do grupy tzw. zespołów czaszkowo-twarzowych, których cechą wspólną jest zniekształcenie czaszki powodujące jej zwężenie i zmniejszenie pojemności. Innymi jednostkami chorobowymi należącymi do do tej grupy są m.in:
- zespół Aperta;
- zespół Pfeiffera;
- zespół Robertsa.
Źródło: https://portalzdrowiadziecka.pl/wp-content/uploads/2016/08/Czaszka-szwy.jpeg
Zespół Crouzona – objawy

Wygląd chorego z zespołem Crouzona jest charakterystyczny – w pierwszej kolejności uwagę zwraca wysokie, wypukłe czoło i hipoplazja (niedorozwój) środkowej części twarzy. Najczęściej przedwczesne zarośnięcie dotyczy szwu wieńcowego, przebiegającego obustronnie pomiędzy kośćmi czołowymi, a ciemieniowymi oraz szwów kości twarzy. Należy jednak pamiętać, że przedwczesne skostnienie może dotknąć każdego ze szwów czaszkowych. Do typowych dla zespołu Crouzona objawów należą:
- hiperteloryzm – szerokie rozstawienie oczodołów i gałek ocznych;
- wytrzeszcz – uwypuklenie gałek ocznych spowodowane obecnością płytkich oczodołów. Wytrzeszcz może prowadzić do przewlekłych zapaleń spojówek i rogówki (w wyniku niedomykania się powiek);
- zapadnięty nos o wyglądzie “dzioba papuziego”;
- deformacje czaszki – wieżowaty, trójkątny lub łódkowaty kształt czaszki w zależności od tego, które szwy czaszkowe uległy przedwczesnemu zrośnięciu;
- zniekształcenie części twarzowej czaszki – “twarz żabia” z niedorozwojem szczęki, kości jarzmowej i wysunięciem żuchwy;
- “gotyckie” podniebienie – podniebienie twarde jest nadmiernie wysklepione i wąskie;
- profil twarzy wyraźnie wklęsły;
- niedomykanie ust;
- wady wzroku, zez zbieżny;
- wady zgryzu – przodożuchwie, stłoczenia zębów;
- niedosłuch wynikający z niedorozwoju zewnętrznych przewodów słuchowych, zaburzeń rozwoju kosteczek słuchowych i trąbki słuchowej.
Rozpowszechnienie i nasilenie wyżej wymienionych objawów jest wśród chorych bardzo zróżnicowane. Łagodne przypadki zespołu Crouzona wykazują zwykle prawidłowy poziom inteligencji, jednak w cięższym przebiegu choroby często dochodzi do zaburzeń rozwoju psychofizycznego dziecka. Wygląd dzieci chorych na zespół Crouzona znacznie odbiega od ich zdrowych rówieśników-dopóki są małe, nie zwracają na to uwagi, lecz wraz z dojrzewaniem poczucie “bycia innym” i odtrącenia przez rówieśników narasta.
Osoby cierpiące na zespół Crouzona o ciężkim przebiegu, w którym kraniosynostoza dotyczy wszystkich szwów czaszkowych, mogą mieć także problemy z mową, połykaniem oraz oddychaniem. Dysfunkcje te wynikają z deformacji i nieprawidłowego ukształtowania kości i elementów chrzęstnych twarzoczaszki (skrzywienie przegrody nosa, zarośnięcie nozdrzy tylnych) oraz poważnych wad zgryzu.
Równie, a nawet bardziej istotnym niż deformacje zewnętrzne problemem jest nadciśnienie śródczaszkowe, powstające u niektórych chorych z powodu braku miejsca w jamie czaszki. Ze względu na kraniosynostozę, kości czaszki nie mogą rosnąć, a mózg stale powiększa swoje rozmiary – z powodu tych dysproporcji wewnątrz czaszki tworzy się nadciśnienie, które jest stanem bardzo niebezpiecznym, zagrażającym życiu. U dzieci, u których występuje nadciśnienie śródczaszkowe, między 6.a 10. miesiącem życia wykonywana jest operacja polegająca na oddzieleniu i przesunięciu do przodu kości czołowej, aby zrobić “miejsce” dla wciąż rosnącego mózgu. Na szczęście, nadciśnienie śródczaszkowe nie dotyczy każdego pacjenta i występuje stosunkowo rzadko, głównie wtedy, gdy przedwczesnemu zrośnięciu uległy wszystkie szwy czaszkowe.
Zespół Crouzona – przyczyny
Przyczyną występowania zespołu Crouzona są mutacje w genie FGFR2- gen ten zawiera “instrukcje” dotyczące wytwarzania białka zwanego receptorem czynnika fibroblastów 2. Fibroblasty są to komórki tkanki łącznej, mające zdolność do wytwarzania substancji międzykomórkowej składającej się z włókien kolagenowych. Białko będące produktem genu FGFR2 w warunkach fizjologicznych sygnalizuje niedojrzałym komórkom prekusorowym, aby te przekształciły się do komórek kostnych. Uważa się, że mutacje w genie FGFR2 obecne w zespole Crouzona powodują wytwarzanie wyżej wymienionego białka z “nadaktywną sygnalizacją”- skutkuje to przedwczesną transformacją komórek prekursorowych w komórki kostne i zarośnięciem szwów czaszkowych zbyt wcześnie.
Zapamiętaj:
Zespół Crouzona należy do grupy tzw. zespołów czaszkowo-twarzowych, których cechą wspólną jest zniekształcenie czaszki powodujące jej zwężenie i zmniejszenie pojemności.
Zaburzenie to dziedziczone jest w sposób autosomalny dominujący, co oznacza, że do jego wystąpienia wystarczające jest odziedziczenie zaledwie jednej kopii zmutowanego genu,od jednego z rodziców. W większości przypadków (ok. 60%) dziecko dziedziczy zmutowany gen od chorego na zespół Crouzona rodzica i wówczas występuje u niego postać rodzinna choroby. Rzadziej zdarza się, że rodzice nie wykazują cech choroby, a do mutacji w genie FGFR2 dochodzi dopiero podczas zapłodnienia, czyli połączenia plemnika i komórki jajowej – taką sytuację określamy mianem mutacji powstałej “de novo”.
Gdy osoba cierpiąca na zespół Crouzona zdecyduje się na posiadanie potomstwa, każde z jej dzieci ma 50% szans na odziedziczenie mutacji.
Zespół Crouzona – diagnostyka
Ustalenie choroby genetycznej, które z reguły występują rzadko w populacji, jest zazwyczaj trudnym i wieloetapowym procesem. Podejrzenie zespołu Crouzona nasuwa lekarzowi badającemu chorego, charakterystyczny wygląd pacjenta- wypukłe czoło, niedorozwój środkowej części twarzy, wytrzeszcz oczu i deformacje czaszki. Pomocne w wysunięciu podejrzenia tego zespołu genetycznego jest to, że zazwyczaj jedno z rodziców także przedstawia fenotyp (wygląd) charakterystyczny dla zespołu Crouzona, co dowodzi, że dziecko mogło odziedziczyć po nim tę chorobę. W diagnostyce różnicowej schorzenia wykorzystywane są także badania radiologiczne, takie jak tomografia komputerowa i rezonans magnetyczny, które obrazują nieprawidłowości w budowie twarzoczaszki, zatok obocznych nosa oraz zarośnięcie szwów czaszkowych. Diagnostyką pacjentów zajmują się pediatrzy we współpracy z genetykami klinicznymi, gdyż ostateczne potwierdzenie rozpoznania zespołu Crouzona możliwe jest jedynie poprzez wykrycie mutacji genu FGFR2 w badaniach genetycznych. W celu wykonania badania genetycznego od pacjenta pobierana jest krew z żyły obwodowej.
Zespół Crouzona – leczenie
Leczenie dzieci z zespołem Crouzona jest procesem multidyscyplinarnym, obejmującym zabiegi operacyjne (często wieloetapowe) oraz opiekę różnych specjalistów m.in. fizjoterapeutów, neurologopedów i psychologów, ze względu na występujące w zespole zaburzenia neurorozwojowe. Leczeniem operacyjnym zespołu Crouzona zajmują się neurochirurdzy, chirurdzy szczękowo- twarzowi oraz chirurdzy plastyczni. W przypadku nasilonych wad zgryzu bardzo istotne jest leczenie ortodontyczne, a przy istniejących powikłaniach ze strony narządu wzroku (zapalenie spojówek, zez, wady wzroku) lub słuchu (niedosłuch) wskazana jest także specjalistyczna opieka okulistyczna i laryngologiczna. Pacjenci cierpiący na niedosłuch powinni zostać zaopatrzeni w aparaty słuchowe.
Czy wiesz że:
w zespole Crouzona nie występują deformacje w obrębie dłoni i stóp, co odróżnia go od innego zaburzenia genetycznego związanego z przedwczesnym zarośnięciem szwów czaszkowych- zespołu Aperta.
Interwencje chirurgiczne mają na celu zapobieganie powikłaniom mózgowym, okulistycznym oraz oddechowym, a także korygowanie deformacji twarzy. Efekt terapeutyczny osiągany jest poprzez nacięcie i wysunięcie ku przodowi elementów kostnych czaszki w taki sposób, aby rozwijający się mózg miał wystarczająco dużo miejsca do zwiększania swojej objętości, a następnie stabilizację kości w nowym miejscu za pomocą metalowych płytek. Po operacji pacjent musi przez kilka miesięcy nosić specjalny hełm, aby na nowo nadać kształt czaszce i chronić ją przed urazami. Podejście chirurgiczne powinno być spersonalizowane, biorąc pod uwagę konkretnego pacjenta i jego problemy zdrowotne.
Wczesne rozpoznanie zespołu Crouzona oraz wdrożenie leczenia chirurgicznego wpływa na zmniejszenie zniekształceń części twarzowej czaszki chorych, a leczenie ortodontyczne zapewnia prawidłową wymowę, oddychanie i połykanie. Leczenie pacjentów z zespołem Crouzona jest wieloletnie i wymaga współpracy wielu specjalistów, lecz tylko takie postępowanie daje nadzieję na ich optymalny rozwój. Choć tak jak inne zaburzenia genetyczne, zespół Crouzona jest na dzień dzisiejszy nieuleczalny, rzetelnie prowadzona opieka wielospecjalistyczna znacznie poprawia komfort życia pacjentów i daje im szansę na długie życie.