

Mianem wad wrodzonych określamy wszelkie patologie, które powstają u płodu podczas ciąży. Szacuje się, że różnego rodzaju wady wrodzone dotyczą 2-5% wszystkich noworodków. Wady te mogą być izolowane lub mogą tworzyć charakterystyczne zestawienia, które określamy jako zespoły wad wrodzonych. Niektóre zespoły wad wrodzonych o podłożu genetycznym są na tyle charakterystyczne, że do postawienia wstępnej diagnozy wystarczające jest wprawne oko doświadczonego pediatry. Do takich schorzeń zaliczamy zespół Kabuki – skąd pochodzi nazwa tej rzadkiej choroby oraz jak się ona objawia?
Co to jest zespół Kabuki?
Zespół Kabuki, znany także jako zespół Niikawa-Kuroki, jest to rzadkie schorzenie genetyczne, obejmujące zespół wad wrodzonych, którym często towarzyszy niepełnosprawność intelektualna. Schorzenie to po raz pierwszy zostało opisane w 1981 roku przez dwóch japońskich lekarzy – N. Niikawa i Y. Kuroki i początkowo uznawano je za specyficzne dla obszaru Japonii. Współcześnie przypadki zespołu Kabuki opisywane są na całym świecie, ale jego dokładne rozpowszechnienie wśród rasy kaukaskiej nie jest znane. Szacuje się, że schorzenie to występuje z częstością 1:32 000 – 1:86 000 żywych urodzeń w populacji Japonii. Nazwa zespołu pochodzi od tradycyjnego japońskiego teatru Kabuki, którego aktorzy nosili makijaż podobny do specyficznych cech fenotypowych twarzy pacjentów.
Objawy zespołu Kabuki

Najbardziej charakterystycznymi cechami, umożliwiającymi wstępne postawienie rozpoznania zespołu Kabuki, są specyficzne rysy twarzy pacjenta, które przypominają makijaż aktorów występujących w tradycyjnym japońskim teatrze Kabuki. Do cech typowych dla tego schorzenia należą:
- długie szpary powiekowe;
- długie i gęste rzęsy;
- wywinięcie bocznej powierzchni powieki dolnej;
- łukowate brwi, przerzedzone w ⅓ bocznej części łuku brwiowego;
- szeroki grzbiet nosa oraz płaski, zapadnięty koniuszek nosa;
- duże, wydatne i odstające uszy;
- wysokie, nadmiernie wysklepione podniebienie (określane jako “gotyckie podniebienie”);
- obniżenie kącików ust;
- anomalie zębów (zaburzenia w okresie ząbkowania, wady zgryzu, szeroko rozstawione zęby, dodatkowe zęby trzonowe).
Zespół wad wrodzonych, które powodują odmienny, specyficzny wygląd chorych określany jest mianem dysmorfii. Zmiany dysmorficzne twarzy obserwowane są niemal u wszystkich pacjentów z zespołem Kabuki. Rzadko stwierdza się ponadto rozszczep wargi i/lub podniebienia, niebieskie zabarwienie twardówek, opadanie powiek lub zeza.
Różnego stopnia dysmorfia twarzy u pacjentów z zespołem Kabuki
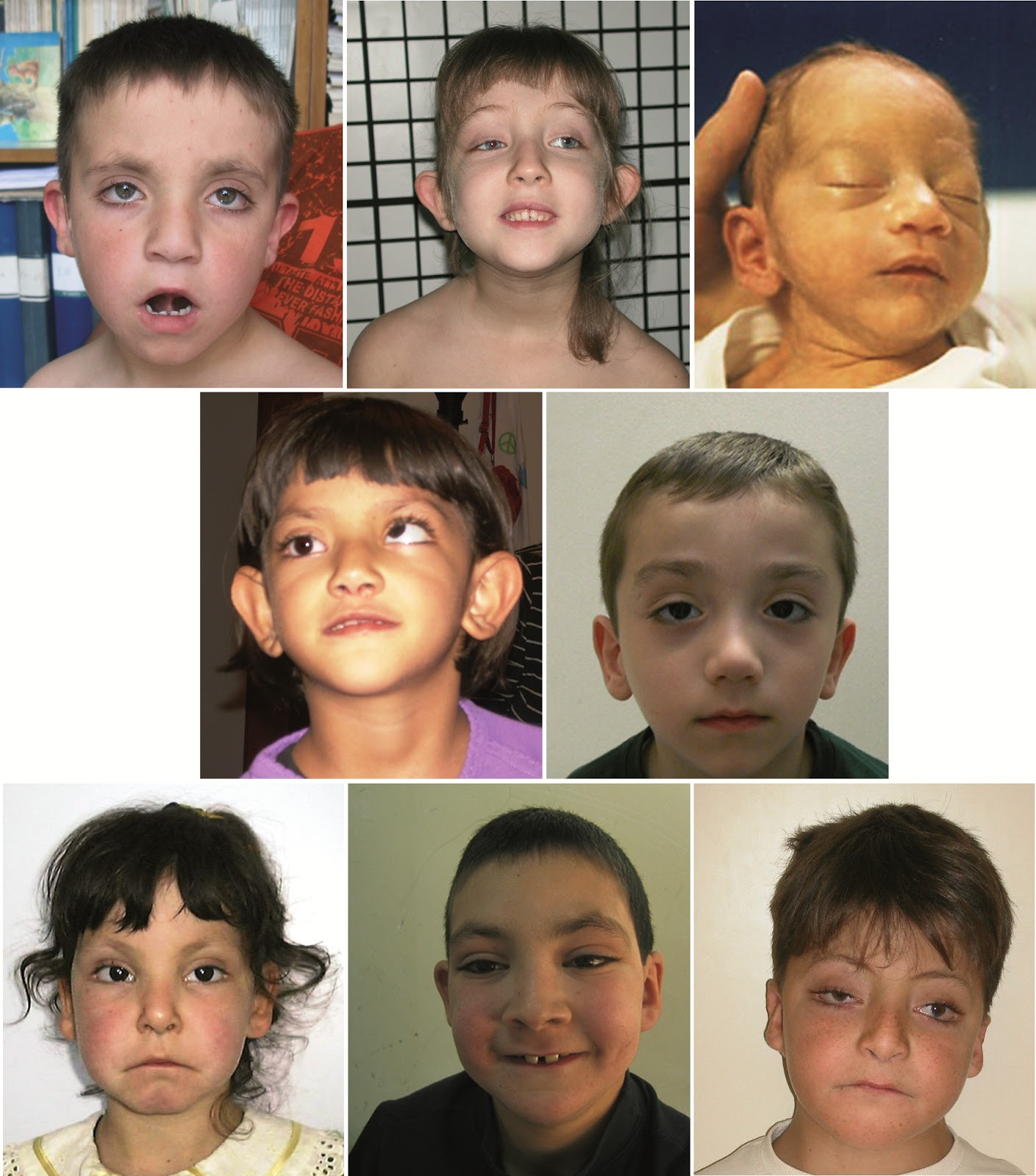
Poza charakterystycznym wyglądem twarzy, pacjenci z zespołem Kabuki prezentują także szereg zaburzeń rozwojowych, neurologicznych oraz wad narządów wewnętrznych, do których zaliczamy:
- niepełnosprawność intelektualną w stopniu łagodnym do umiarkowanego (większość pacjentów jest w stanie werbalnie porozumiewać się z otoczeniem i uczęszczać do szkoły specjalnej);
- opóźnienie wzrastania i rozwoju psychoruchowego – pacjenci później niż ich rówieśnicy uczą się siedzieć, chodzić, mówić;
- występowanie napadów drgawek;
- obniżenie napięcia mięśniowego (hipotonia), które w okresie noworodkowym powoduje trudności z karmieniem – noworodki nie wykształcają prawidłowego odruchu ssania pokarmu;
- nieprawidłowości układu kostno-szkieletowego (skolioza, zniekształcenia kręgów, anomalie żeber, brachydaktylia ze skróceniem paliczka środkowego piątego palca, nadmierna ruchomość stawów);
- małogłowie – obwód głowy jest zbyt mały w stosunku do wartości referencyjnych przyjętych dla płci i wieku dziecka;
- przetrwałe “opuszki płodowe” palców dłoni i stóp – bardzo wypukłe opuszki palców, które fizjologicznie występują u człowieka jedynie w okresie płodowym, a następnie zanikają;
- niedosłuch;
- wady układu krążenia (np. ubytek przegrody międzykomorowej lub międzyprzedsionkowej serca) i układu moczowo-płciowego (wady nerek, wnętrostwo, spodziectwo);
- wady przewodu pokarmowego np. zarośnięcie dwunastnicy lub odbytu;
Pacjenci z zespołem Kabuki wykazują zwiększoną podatność na infekcje oraz częściej niż zdrowi rówieśnicy zapadają na choroby autoimmunologiczne (np. chorobę Hashimoto, niedoczynność nadnerczy). Typowe dla tego schorzenia jest również występowanie przedwczesnego dojrzewania, które stwierdzane jest, jeżeli jego objawy występują przed 8 rokiem życia u dziewcząt i przed 9 rokiem życia u chłopców. Przedwczesne dojrzewanie częściej stwierdzane jest u dziewcząt (przedwczesny rozwój piersi i pierwsza miesiączka).
*twardówka – zewnętrzna powierzchnia gałki ocznej, nadająca oku kształt
*skolioza – boczne skrzywienie kręgosłupa
*brachydaktylia – wada wrodzona polegająca na skróceniu palców
*wnętrostwo – niezstąpienie jąder do worka mosznowego
*spodziectwo – wada wrodzona dotyczącą układu moczowo-płciowego, która występuje u chłopców i polega na nieprawidłowej lokalizacji zewnętrznego ujścia cewki moczowej (znajduje się ono na brzusznej powierzchni prącia zamiast na żołędziu).
Przyczyny zespołu Kabuki
Przyczyną występowania zespołu Kabuki jest mutacja genu KMTD2, który jest zlokalizowany na chromosomie 12 (55-80% przypadków) lub genu KDM6A, zlokalizowanego na chromosomie płciowym X (2-6% przypadków). Dokładna funkcja obu genów nie została jeszcze poznana, ale uważa się, że odpowiadają one za kontrolę metylacji histonów, czyli białek strukturalnych wiążących DNA i nadających kształt chromosomom. Tym samym, powyższe geny wpływają na aktywację odpowiednich genów podczas rozwoju organizmu. Mutacje prowadzą do zaburzeń metylacji histonów, przez co aktywacja określonych genów jest zaburzona. Skutkiem tego jest powstawanie wad rozwojowych, charakterystycznych dla zespołu Kabuki. U pacjentów z mutacją genu KMTD2 częściej występują dysfunkcje nerek, nadmierna ruchomość stawów, wady podniebienia i przedwczesne dojrzewanie płciowe. Chorzy z mutacją genu KDM6A częściej cierpią na nadmierny porost włosów, anomalie w budowie zębów oraz palców.
Czy wiesz że:
Kabuki to tradycyjny rodzaj teatru japońskiego wywodzący się z XVII wieku, który został wpisany na listę niematerialnego dziedzictwa UNESCO?
W znacznej większości przypadków zespół Kabuki nie jest dziedziczny – powstaje na skutek mutacji de novo, która w 55-80% przypadków dotyczy genu KMTD2. Termin “de novo” oznacza, że mutacja nie występuje u rodziców pacjenta, lecz dochodzi do niej podczas zapłodnienia, czyli połączenia plemnika i komórki jajowej. Opisywano także rodzinne występowanie zespołu Kabuki o mechanizmie dziedziczenia autosomalnym dominującym. W takiej sytuacji, zmutowany gen KMTD2 występuje u jednego rodziców i może on przekazać go potomstwu z 50% prawdopodobieństwem. Odziedziczenie jednej kopii zmutowanego genu (od jednego z rodziców) jest wystarczające do ujawnienia się objawów choroby u dziecka.
Mutacje genu KDM6A jedynie w 2-6% są odpowiedzialne za powstawanie zespołu Kabuki i najczęściej są to mutacje de novo (nieodziedziczone od rodziców, powstające spontanicznie podczas zapłodnienia lub na wczesnym etapie życia płodowego). U nielicznych pacjentów, mutacje genu KDM6A mogą być dziedziczone w sprzężeniu z chromosomem X (gen ten zlokalizowany jest na chromosomie płciowym X). Kobiety mają dwa chromosomy płciowe X (kariotyp żeński to 46, XX), a mężczyźni – jeden chromosom X i jeden chromosom Y ( kariotyp męski to 46, XY). Z tego powodu, jeżeli kobieta odziedziczy wadliwą kopię genu KDM6A od rodzica, objawy zespołu Kabuki będą u niej łagodne, ponieważ posiada ona drugą, prawidłową kopię tego genu na drugim chromosomie płciowym X. W przypadku mężczyzn odziedziczenie jednej wadliwej kopii genu jest wystarczające do rozwoju pełnoobjawowej choroby, ponieważ mają oni tylko jeden chromosom płciowy X – nie posiadają drugiej, prawidłowej kopii genu, która mogłaby zrównoważyć obecność zmienionej kopii (chromosom Y zawiera znacznie mniej genów).
U około 20% pacjentów przyczyna występowania zespołu Kabuki pozostaje nieznana – nie udaje się jednoznacznie ustalić genetycznego podłoża choroby (nie są oni nosicielami wyżej wymienionych zmian genetycznych).
*metylacja – proces polegający na przyłączaniu grup metylowych do nukleotydów (podstawowych jednostek budujących DNA), który zachodzi przy udziale enzymów – metylaz.
*kariotyp – zestaw chromosomów charakterystyczny dla danego organizmu. Na kariotyp człowieka składają się 22 pary chromosomów autosomalnych (odpowiadających za dziedziczenie wszystkich cech organizmu z wyjątkiem cech związanych z płcią) i jedna para chromosomów płciowych.
Diagnostyka zespołu Kabuki
Zespół Kabuki należy podejrzewać u osób z:
- jakąkolwiek kombinacją pięciu głównych objawów zdefiniowanych przez doktora Niikawa i współpracowników;
- obecnością niżej wymienionych anomalii strukturalnych lub nieprawidłowości funkcjonalnych.
Główne objawy:
- typowe rysy twarzy;
- anomalie układu kostno – szkieletowego;
- nieprawidłowości skórne – przetrwałe “opuszki płodowe” palców dłoni i stóp;
- niepełnosprawność intelektualna w stopniu lekkim do umiarkowanego;
- opóźnienie wzrastania.
Anomalie strukturalne:
- nieprawidłowości dotyczące narządu wzroku (opadanie powiek, zez);
- rozszczep wargi i/lub podniebienia;
- wady zębów;
- wrodzone wady serca;
- wady przewodu pokarmowego, w tym zarośnięcie odbytu;
- anomalie układu moczowo-płciowego, w tym wnętrostwo u mężczyzn.
Nieprawidłowości funkcjonalne:
- niedosłuch;
- trudności z karmieniem;
- nieprawidłowości endokrynologiczne, w tym przedwczesne dojrzewanie płciowe;
- zwiększona zapadalność na różnego rodzaju infekcje i schorzenia autoimmunologiczne;
- występowanie drgawek.
Pewne rozpoznanie zespołu Kabuki można postawić u pacjenta z historią występowania hipotonii w dzieciństwie, opóźnieniem rozwoju psychoruchowego i/lub wykazującego cechy niepełnosprawności intelektualnej, który spełnił ≥1 z poniższych kryteriów:
a). obecność typowych cech dysmorficznych – długich szpar powiekowych z wywinięciem bocznej ⅓ dolnej powieki oraz ≥2 z poniższych:
- łukowate brwi, przerzedzone w bocznej części,
- płaski, zapadnięty koniuszek nosa,
- duże, wydatne uszy,
- przetrwałe “opuszki płodowe” palców dłoni i stóp;
b). potwierdzona mutacja w genie KTMD2 lub KDM6A.
Jak leczyć zespół Kabuki?
Zespół Kabuki jest schorzeniem o podłożu genetycznym i na dzień dzisiejszy nie opracowano metody skutecznego leczenia przyczynowego. Podstawą terapii pozostaje spersonalizowane leczenie objawowe, czyli niwelowanie dokuczliwych objawów występujących u danego pacjenta.
Chorzy z rozpoznanym zespołem Kabuki wymagają opieki multidyscyplinarnego zespołu przez całe życie. Powinni regularnie uczęszczać na wizyty kontrolne do genetyka klinicznego, pediatry, psychologa, neurologopedy, fizjoterapeuty, okulisty oraz do lekarzy innych specjalności, w zależności od problemów zdrowotnych dotyczących konkretnego pacjenta. Przykładowo, występowanie napadów padaczkowych wymaga stałej opieki neurologicznej i przyjmowania leków przeciwpadaczkowych, wrodzona wada serca powinna być leczona operacyjnie przez kardiochirurga, a wady układu moczowo-płciowego wymagają leczenia urologicznego. Wszelkie zaburzenia endokrynologiczne, w tym przedwczesne dojrzewanie płciowe, powinny być diagnozowane i leczone przez endokrynologa. W razie nieprawidłowych wyników badań immunologicznych lub nawracających infekcji, zalecana jest konsultacja z immunologiem klinicznym. Regularna ocena rozwoju pacjenta przez specjalistów pozwala na dostosowanie terapii do indywidualnych potrzeb chorego.
Zapamiętaj:
Nazwa zespołu pochodzi od tradycyjnego japońskiego teatru Kabuki, którego aktorzy nosili makijaż podobny do specyficznych cech fenotypowych twarzy pacjentów.
Jak już wspomniano, częstym problemem zdrowotnym u pacjentów z zespołem Kabuki są różnego rodzaju nieprawidłowości w obrębie układu kostno-stawowego, w tym nadmierna ruchomość stawów. Z tego względu chorzy powinni pozostawać pod stałą opieki ortopedy i fizjoterapeuty, gdyż jedynie regularna rehabilitacja daje szanse na utrzymanie sprawności. Pacjenci z nadmierną wiotkością stawów powinni unikać czynności zwiększających ryzyko urazów, jak np. skakanie na trampolinie. Dzieci, u których występują trudności ze ssaniem, mogą wymagać karmienia przez zgłębnik nosowo-żołądkowy. Jest to cienka, giętka rurka wprowadzana przez nos, a następnie przechodząca przez tylną ścianę gardła i przełyk wprost do żołądka pacjenta. Do zgłębnika podaje się płynny pokarm przy pomocy specjalnej strzykawki.
Rokowanie w przebiegu zespołu Kabuki jest korzystne – nie wykazano, aby schorzenie to istotnie wpływało na długość życia pacjentów. Większość problemów zdrowotnych pojawia się już w okresie dziecięcym i mogą być one skutecznie leczone, tak jak np. wady wrodzone serca. Długość życia w największym stopniu uzależniona jest od obciążeń kardiologicznych i immunologicznych (nawracających infekcji), występujących u konkretnego chorego.
Zespół Kabuki jest to schorzenie o podłożu genetycznym, którego nazwa wywodzi się od charakterystycznych rysów twarzy pacjentów, przypominających makijaż aktorów występujących w teatrze Kabuki. Stosunkowo rzadkie występowanie zespołu Kabuki, jak również szerokie spektrum objawów klinicznych i zróżnicowane nasilenie choroby u poszczególnych pacjentów powodują trudności we właściwej diagnostyce zespołu. Aby rozpowszechniać wiedzę na temat tej mało znanej i wciąż nieuleczalnej choroby, dzień 23 października ustanowiono Dniem Zespołu Kabuki.